近日,北京化工大学程道建教授课题组等人设计出了一种二硫化钼(MoS2)和磷化钼(MoP)结合的复合材料,其层间界面结构有较高的活性位点,能有效缓解商业化催化剂在碱性下析氢反应(HER)的不足。该研究成果已被发表在国际著名能源期刊Nano Energy上。

目前,电解水技术被认为是一种大规模生产氢气的绿色清洁方法,而HER在氢气制备过程中起着至关重要的作用。然而,HER在碱性电解液中的反应动力学十分缓慢,就连当前最先进的催化剂——贵金属铂族金属(PGM)在其中的动力学比在酸性条件下的动力学慢两个数量级,这严重制约了基于碱性条件下水分解的制氢技术的发展。
MoS2因有活性高和稳定性优异的特点,而被公认为是PGM催化剂的理想替代品。电化学实验和密度泛函理论(DFT)计算表明,MoS2在酸性溶液中表现出优异的HER性能,因为二硫化钼的边缘位置的最佳氢吸附自由能与铂的非常接近,约为0.08eV。但是,MoS2在碱性溶液中的HER动力学表现也不是很理想。因此,为了加速水分解的步骤,向MoS2催化剂中引入具有裂解HO-H键功能的“促进剂”,如碳、氮、磷、硒等。
此外,过渡金属MoP由于具有与MoS2相似的结构,而能在碱性条件下表现出更好的HER性能。因此,若将MoP与MoS2催化剂结合,或能起到协同促进碱性介质中的HER动力学的作用。
北京化工大学研究者通过水热法和高温部分磷化手段,在碳布上合成了MoS2-MoP/NC异质结催化剂,其碱性条件下表现出超越商业Pt/C的催化性能,并且有望取代Pt/C催化剂。当电流密度为10mA/cm2时,MoS2-MoP/NC异质结催化剂所需电势仅34 mV,远低于其他材料。结合实验表征与第一性原理计算,表明MoS2-MoP/NC增强的碱性HER催化性能的起源来自通过层间界面结构所引入新的高效活性位点。
然而,研究者在做实验过程中并未一帆风顺。为了解决MoS2催化剂的活性位点数量少,导电性以及稳定性不良等问题,他们向MoS2中引入新的杂原子(P),形成异质界面,界面效应使得界面处S空位增加,从而提高了催化剂边缘活性位点数量。此外,引入导电性优异的碳物质,能有效的提高MoS2基催化剂导电性。最后,通过构建异质结三维催化剂,有利于反应物的吸附以及产物的迅速脱附,大大地提高了催化剂电化学稳定性。
首先根据图1, 通过一系列的水热及高温磷化等步骤便可得到MoS2/NC@CC。

图1.碳布上合成MoS2-MoP/NC示意图
通过图2中SEM、TEM电镜看出碳布表面均匀分散了一层活性物质,活性物质表面包覆了一层碳层,而XRD则证明碳布上同时存在的物质为MoS2和MoP的,证明磷化后部分MoS2转化为MoP。此外,如图2g和图片2h所示,HRTEM验证了MoS2-MoP异质结的存在,界面的构建对于活性的提高将起到关键作用。最后,图片2i表明了Mo、S、P、C、N元素在催化剂材料表面均匀的分布。
如图3所示,根据XPS表征结果,由Mo,S,P谱图中可以看出,该异质结催化剂与对比样品相比,峰的位置发生明显偏移,表明该异质结催化剂中存在电子转移,即有界面效应的存在。此外,从S 2p的谱图中可以看出,该异质结催化剂中,有大量S空位的存在,从C以及N 的谱图中可以看出,该异质结催化剂成功引入碳物质,这有助于提高催化剂的导电性。EPR谱图同样验证MoS2-MoP/NC拥有大量的S空位,这应该是由MoS2-MoP之间的界面导致的。从拉曼光谱可以看出磷化后的材料MoS2的层数小于原始MoS2层数,这是由于磷化过程中部分MoS2层转化为MoP导致的。

图2.The(a-c)MoS2-MoP/NC@CC在不同倍率下的SEM图。(d,e)MoS2-MoP/NC的TEM 和 (g, h)HRTEM图像。(f)MoS2-MoP/NC@CC的XRD结果。(i)MoS2-MoP/NC中相应的Mo, S, P, C, N元素的mapping分布图。

图3.(a)XPS测量光谱。MoS2-MoP/NC中(b) Mo 3d,(c)S 2p,(d) P 2p,(e)C 1s and (f)N 1s的高分辨XPS分析图谱。

图4.(a)MoS2-MoP/NC,MoS2/NC,MoP/NC,Pt/C和纯碳纸在1M KOH溶液中扫描速率为2mV s−1时的LSV曲线(具有iR补偿)。(b) MoS2-MoP/NC与对比样品在电流密度为10和100 mA cm−2条件下性能比较。(c)催化剂对应塔菲尔斜率。(d)在电位0.15V vs. RHE下MoS2-MoP/NC, MoS2/NC,MoP/NC的双电层电容拟合。
根据图4,MoS2-MoP/NC表现出超越商业Pt/C的催化性能,电流密度为10 mA cm-2时,所需电势仅为34mV。远低于其他对比材料。在电流密度超过62mA/cm2时,拥有超越商业Pt/C的催化活性。其对应的Tafel斜率为30mV/dec。根据图5,电化学阻抗谱图表明MoS2-MoP/NC导电性良好,有助于电解水析氢电极上电子的传递,进而促进催化活性的提高。经过10000圈长循环、72小时变电势稳定性测试后,MoS2-MoP/NC催化活性没有明显衰退。
最后,由图6和图7得到,密度泛函理论(DFT)计算证明,MoS2和MoP之间的层间界面的构建有助于MoS2基面上S空位的形成。由于S空位内部电子的离域,MoS2暴露的不饱和配位的Mo原子为水的吸附和解离提供了活性位点,从而加速了水的离解步骤。另一方面,通过层间界面的范德华力减弱MoP对氢的吸附,从而促进了氢气的形成和解吸。因此,我们的结果表明MoS2-MoP/NC增强的碱性HER催化性能的起源来自通过层间界面结构所引入新的高效活性位点。
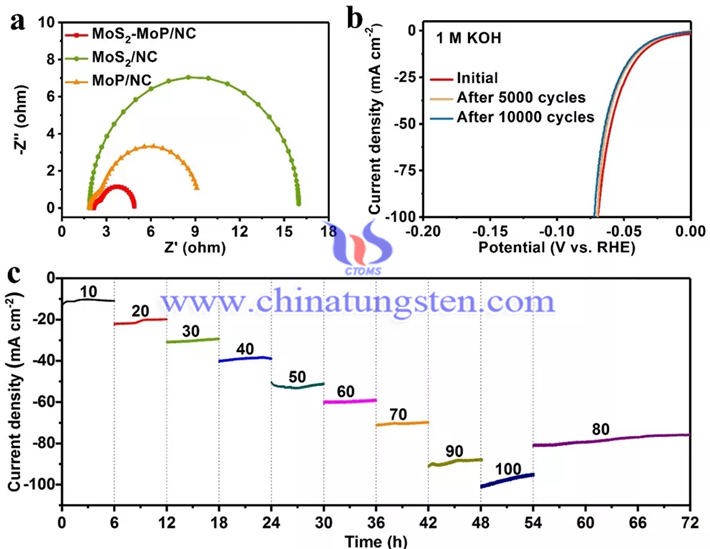
图5.(a) MoS2−MoP/NC,MoS2/NC和MoP/NC在1 M KOH溶液中阻抗谱。(b) MoS2−MoP/NC在5000和10000 CV循环后的极化曲线稳定性测试图。(c) HER在不同电流密度下的计时电流测试(iR补偿)。
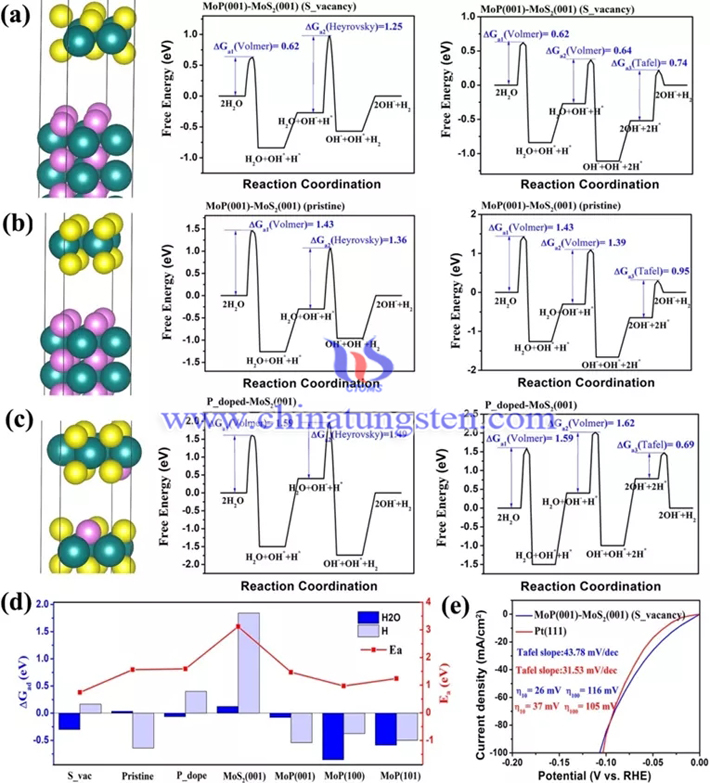
图6.(a)MoP(001)@MoS2(001)-S-vacancy模型图及相应的碱性HER过程的自由能变化。(b)MoP(001)@MoS2(001)-pristine模型图及相应的碱性HER过程的自由能变化。(c)P掺杂MoS2(001)模型图及相应的碱性HER过程的自由能变化。青色、粉红色和黄色分别代表钼、磷和硫原子(d)各种催化剂的H2O吸附自由能、氢吸附自由能(ΔGadH2O,ΔGadH)和碱性HER速控步活化能(Ea)的比较。(e)在转速为1600转/分钟时,Pt(111)与MoP(001)@MoS2(001)-S_vacancy的模拟电流-电位极化曲线的比较。

图7.(a)MoP(001)@MoS2(001)-S_vacancy,MoS2(001)和MoP(100)沿着以(001)为法线的切面的电子限域函数。(b)H2O吸附的电荷密度差和电子转移。蓝色代表失电子,红色代表得电子。(c)被吸附的H2O的氢氧键的投影晶体轨道哈密顿量(COHP)和积分晶体轨道哈密顿量(ICOHP)。
综上所得:(1)提出利用高温磷化法,使MoS2局部磷化,成功地合成出MoS2-MoP/NC界面异质结催化剂;(2)表征与实验证明了MoS2-MoP/NC中界面有S空位存在,进而使之在碱性介质中显示出优于商业Pt/C的HER活性;(3)密度泛函理论(DFT)计算表明,MoS2和MoP之间的界面结构有助于在MoS2的基面上形成S空位,并暴露不饱和配位的Mo原子,这有助于H2O的吸附与裂解;(4)界面间的范德华力可以减弱MoP对氢的吸附,从而促进了氢气的形成和解吸;(5)通过构建MoS2和MoP之间的层间界面,可以成功引入新的高效活性位点,这也是催化性能增强的原因。


